BINF731 - Assignment 2. Protein modeling. .
a) Select a sequence of a human enzyme from one of the protein sequence databases that has a homolog in PDB with at least 30%, but not more than 60% of sequence identity. (Recommended length of the selected sequence is 150-250 residues).
I started by using Uniprot (TrEMBL) database and searching by the parameters of ‘Homo Sapiens’ and sequence length 150-250. To speed up the search I focused on transmembrane proteins. I would then confirm the Uniprot page didn’t have the PDB listed structure as well as PDB directly to confirm it’s of unknown structure. The Human protein selected is Claudin (Gene CLDN1). UniProtKB - A5JSJ9 (A5JSJ9_HUMAN)
>tr|A5JSJ9|A5JSJ9_HUMAN Claudin OS=Homo sapiens OX=9606 GN=CLDN1 PE=2 SV=1 MANAGLQLLGFILAFLGWIGAIVSTALPQWRIYSYAGDNIVTAQAMYEGLWMSCVSQSTG QIQCKVFDSLLNLSSTLQATRALMVVGILLGVIAIFVATVGMKCMKCLEDDEVQKMRMAV IGGAIFLLAGLAILVATAWYGNRIVQEFYDPMTPVNARYEFGQALFTGWAAASLCLLGGA LLCCSCPRKTTSYPTPRPYPKPAPSSGKDYV
Searching PDB using the sequence gives 9 hits ranging from 34% to 60% identity with four belonging to Homo Sapiens source organism. (Default search parameters with ‘Display results as: Polymer entities’ to show sequence identity match%).
b) Predict the three-dimensional structure for the selected sequence using at least two methods, e.g., fold recognition and homology modeling (Phyre2, I-TASSER, RaptorX, SwissModel, Modweb, etc.).
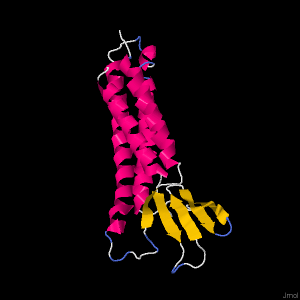
The amino acid sequence was used to predict the three-dimensional structure using Phyre2 and SwissModel.
For Phyre2 the modelling mode was set to ‘normal’ and the following 3D model was produced.While Global model quality estimate (GMQE) is 0.67 +- 0.06 indicating a higher reliability, this quality estimating was estimated from a target-template alignment to reflect the accuracy of the built model. Therefore, I will resort to analyzing the quality through the QMEAN. The QMEAN is -5.05. The QMEAN z-score can used to analyze quality as it consists of other individual terms. A QMEAN score of -4.0 or below indicates our model is of low quality. After brief consultation of the QMEAN publication, this suggests that our model doesn’t agree with experimental structures of similar size. This isn’t too surprising considering it is a transmembrane protein and other known structure proteins are from other Claudin gene classes.
Benkert, P., Biasini, M., Schwede, T. Toward the estimation of the absolute quality of individual protein structure models. Bioinformatics 27, 343-350 (2011).” https://swissmodel.expasy.org/docs/help#GMQE
d) Evaluate separately one of the biologically important parts of the structure (e.g., an active site or binding site)
2.Konrad M, Schaller A, Seelow D, et al. Mutations in the tight-junction gene claudin 19 (CLDN19) are associated with renal magnesium wasting, renal failure, and severe ocular involvement. Am J Hum Genet. 2006;79(5):949-957. doi:10.1086/508617
-Mohammed El Allam
If your browser/OS combination is Java capable, you will get snappier performance if you use Java